Protein structure prediction
Three-dimensional structure of proteins provides a critical clue to understand protein functions and the mechanism of organisms. Although genomes have been currently deciphered in various organisms, three-dimensional structures of many proteins which are translated from genes still remain unknown. For this reason, computational prediction of three-dimensional protein structure is highly anticipated as it requires less time and labor. We are engaged in a research theme to predict the three-dimensional structure of proteins with high accuracy, based on both information accumulated in databases and physics-based analysis.
- Quarterly structure prediction for monomers
- Protein-protein docking prediction
- Structure refinement for protein structure prediction
- Structure evaluation for protein structure prediction
Quarterly structure prediction for monomers
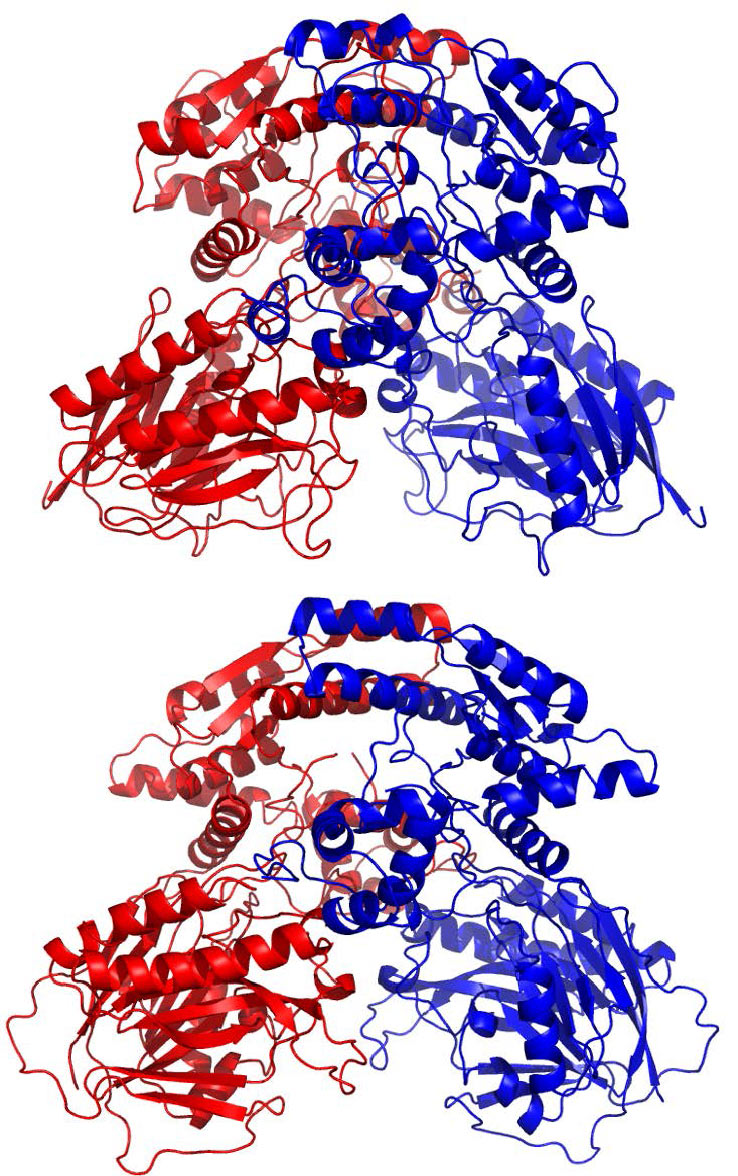
In most cases, proteins form polymers (homomer) where several of the same molecules assemble together. As a result, the homomeric state of proteins needs to be taken into consideration when analyzing the detailed interaction with other molecules. We are currently developing a system which automatically predicts the three-dimensional structure and homomeric state of a protein by entering its amino acid sequence. The figure is a homomeric structure of NAD+ synthetase (590 residues × 2, PDB ID: 3N05) which was predicted from the amino acid sequence using a fully automated server. The top and the bottom show the predicted structure and natural structure, respectively, demonstrating that the three-dimensional structure of each molecule and the relative positioning of the two molecules were successfully predicted with sufficient accuracy.
Structure refinement for protein structure prediction
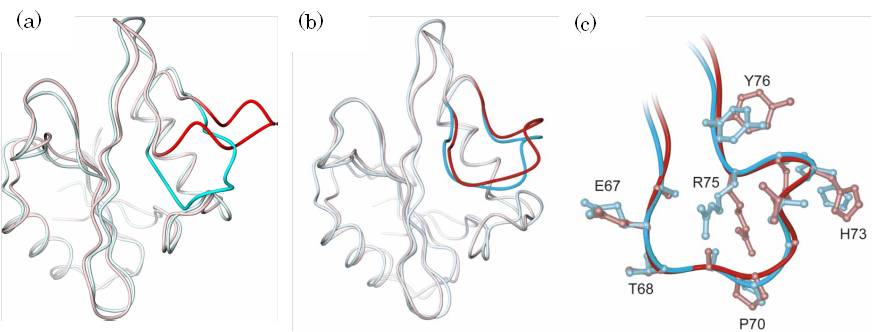
In yet another research project in our group, in vivo molecular structure is predicted using a structure predicted from structure modeling software as an initial structure and applying molecular simulation which takes solvent molecules into consideration. The figure shows a structure of the SH2 domain of serum amyloid P (SAP) component, which was predicted based on the structure of the SH2 domain of the tyrosine kinase P56lck as a template. (a) represents a crystal structure of the SAP protein and the structure was predicted under the assumption that the crystal structure was unavailable. Blue and red show crystal structure and predicted structure, respectively. Regions in the dark color indicate sites where the corresponding template structures were not obtained during structure prediction and show their large deviations from the crystal structure. (b) is a set of representative structures that frequently appeared during simulation, which started from the crystal structure in (a) and a predicted structure (They are shown in corresponding colors). It suggests that the simulation brings the predicted structure closer to the crystal structure and also shows that the structure is stable in solvent. (c) is a partially enlarged view of figure (b).
Structure evaluation for protein structure prediction
A method is under development, which selects a predicted structure from several structures generated from a structure prediction system based on various structural characteristics.