Protein interaction predictions
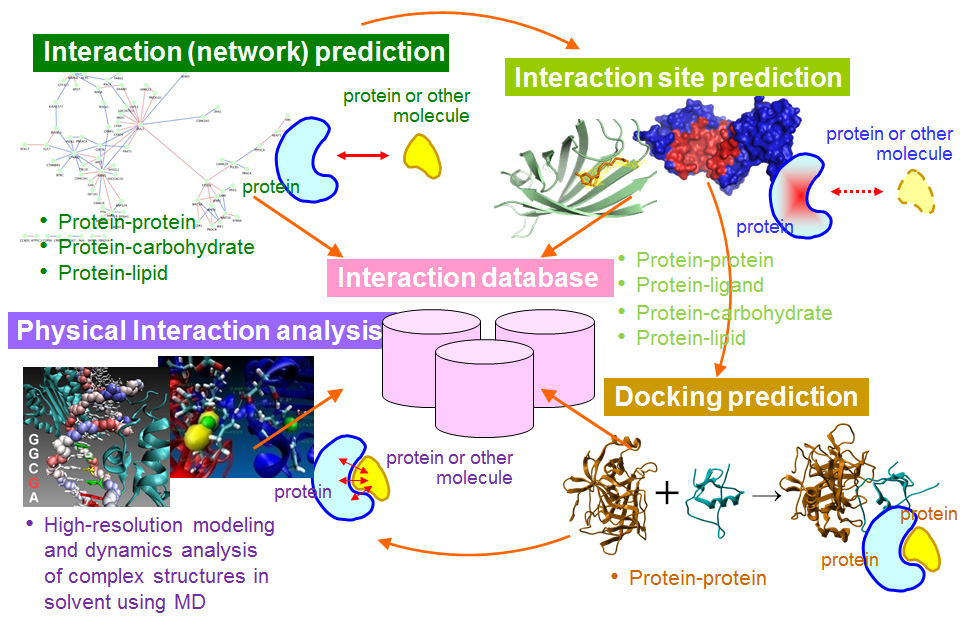
The followings are the three approaches to study protein interactions in our laboratory.
Interaction prediction: To predict whether a given protein interacts with other molecules
Binding-site prediction: To predict residues in a given protein that interact with other molecules and when the structure of a protein is known, to predict the binding site location
Docking prediction: To predict the structure of complexes
We also analyze physical interactions using molecular simulation.
- Synthetic protein binding and binding site prediction system
- Protein binding site prediction using machine learning
- Automatic generation of protein-ligand binding site prediction tools
- Structure-based protein-ligand binding site prediction
- Protein-protein docking prediction
Synthetic protein binding and binding site prediction system
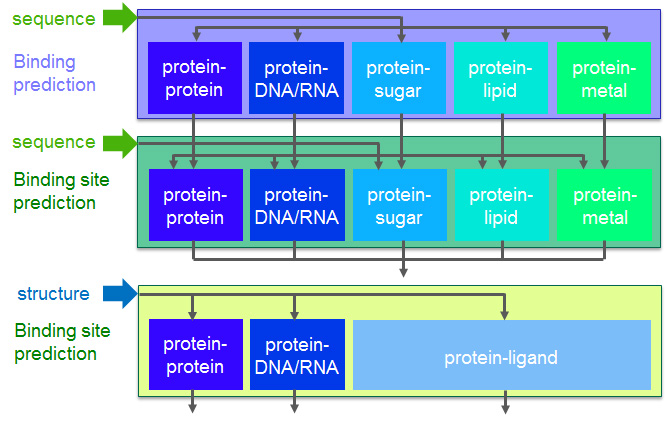
By integrating techniques for protein interaction prediction/binding-site prediction that we have developed or are currently developing, we are constructing a system which enables us to simultaneously predict whether a given protein interacts with proteins, nucleic acids, sugars, lipids, or metals, and if the protein indeed interacts, predict its interaction site.
Protein binding site and functional site prediction using machine learning
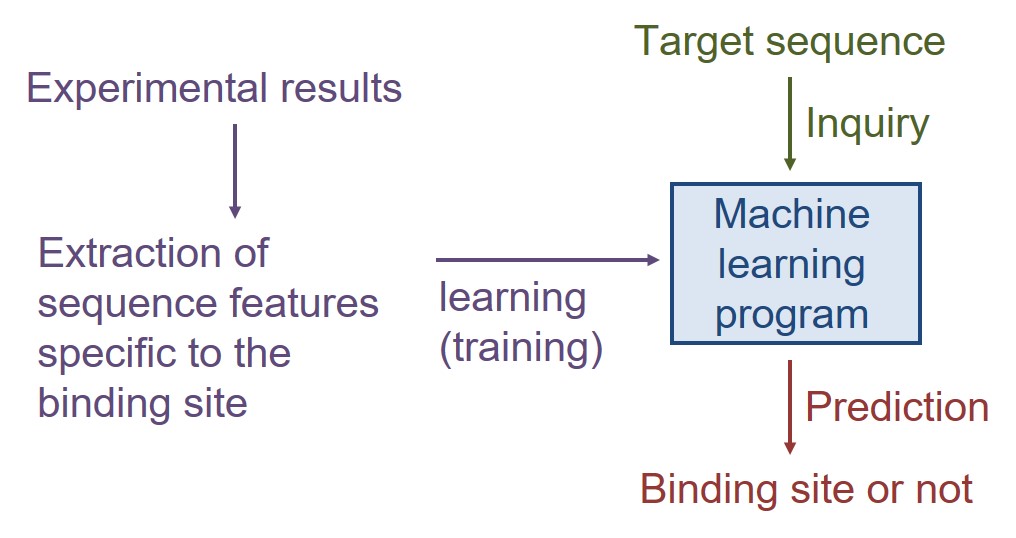
We are currently developing a machine learning-based techniques to predict the interaction and functional site from protein sequences.
Automatic generation of protein-ligand binding site prediction tools
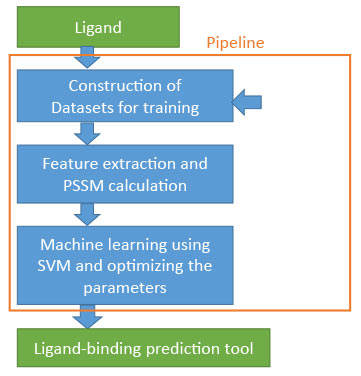
We are also involved in the development of a system which automatically generates tools to predict binding sites for designated ligands. The system automatically collects binding sites of the ligands from structures deposited in PDB, generates their multiple sequence alignments, and calculates the PSSM. This PSSM is entered into the machine learning program SVM to predict ligand binding sites.
At the present moment, the prediction tool can be easily generated within a half- or a full-day. Moreover, this system is also applicable as a basis for further analysis and improvement. The generated tools are reusable as they can be used in the integrated interface.
Structure-based protein-ligand binding site prediction
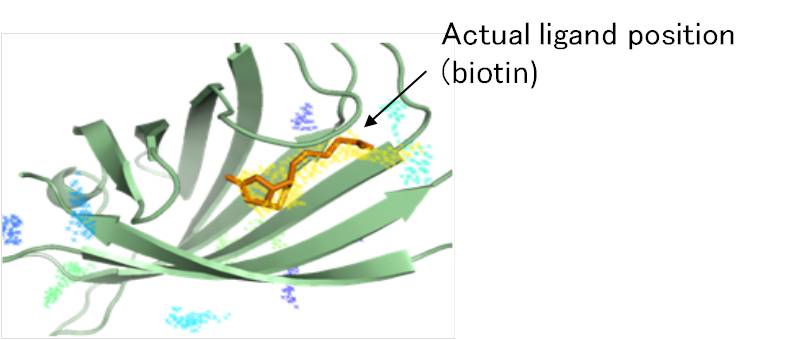
We are also engaged in developing a technique which predicts the site where low molecular weight ligands bind proteins and when proteins bind to low molecular weight ligands. Numerous probes are positioned around a protein to obtain a cluster of probes which have low interaction energies with the protein; the sequence conservation is also taken into account during this step.
The figure is a result which shows the predicted binding site in streptavidin for its ligand, biotin. Among the probes positioned in a grid around the protein structure, the dots represent clustered probes which have low interaction energy with the protein. The prediction order increases from blue to yellow and, in the figure, the predicted site with the highest ranking (yellow) is shown to agree with the actual biotin binding site.
Protein-protein docking prediction
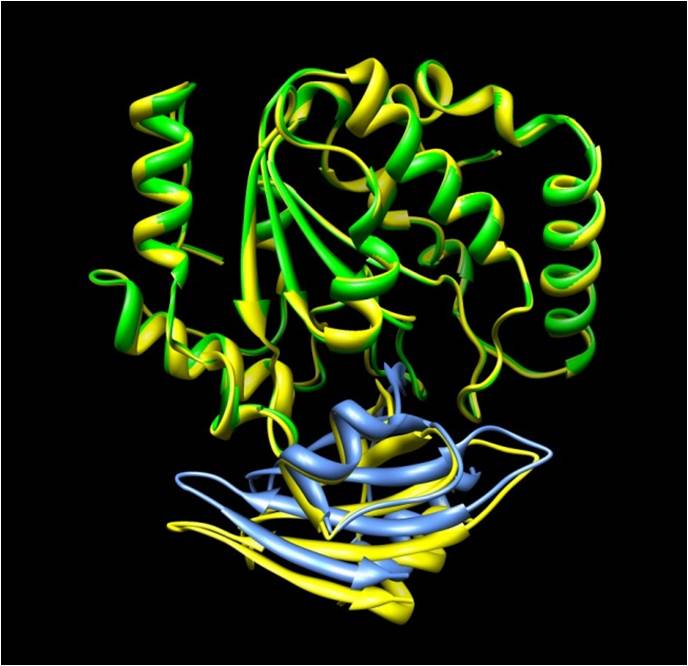
As for docking simulation, we are currently developing a high-speed calculation technique using spherical surface harmonics and the radial basis function. We are attempting to further accelerate the process by dividing a protein molecule into several layers, defining the radial basis function for each layer, and controlling the expansion coefficient. Such docking acceleration is considered to help realize flexible docking by ensemble docking, and be of benefit in comprehensive interaction prediction and substrate exploration.
The figure shows a predicted docking site of glycosylase (green) and its inhibitor (blue). Shown in yellow is a known crystal structure of a complex, superimposed on the prediction result.